


Glioma Subtype Classification & Survival Modeling from DNA Methylation
Classifying brain-tumor subtypes and predicting survival from DNA methylation.
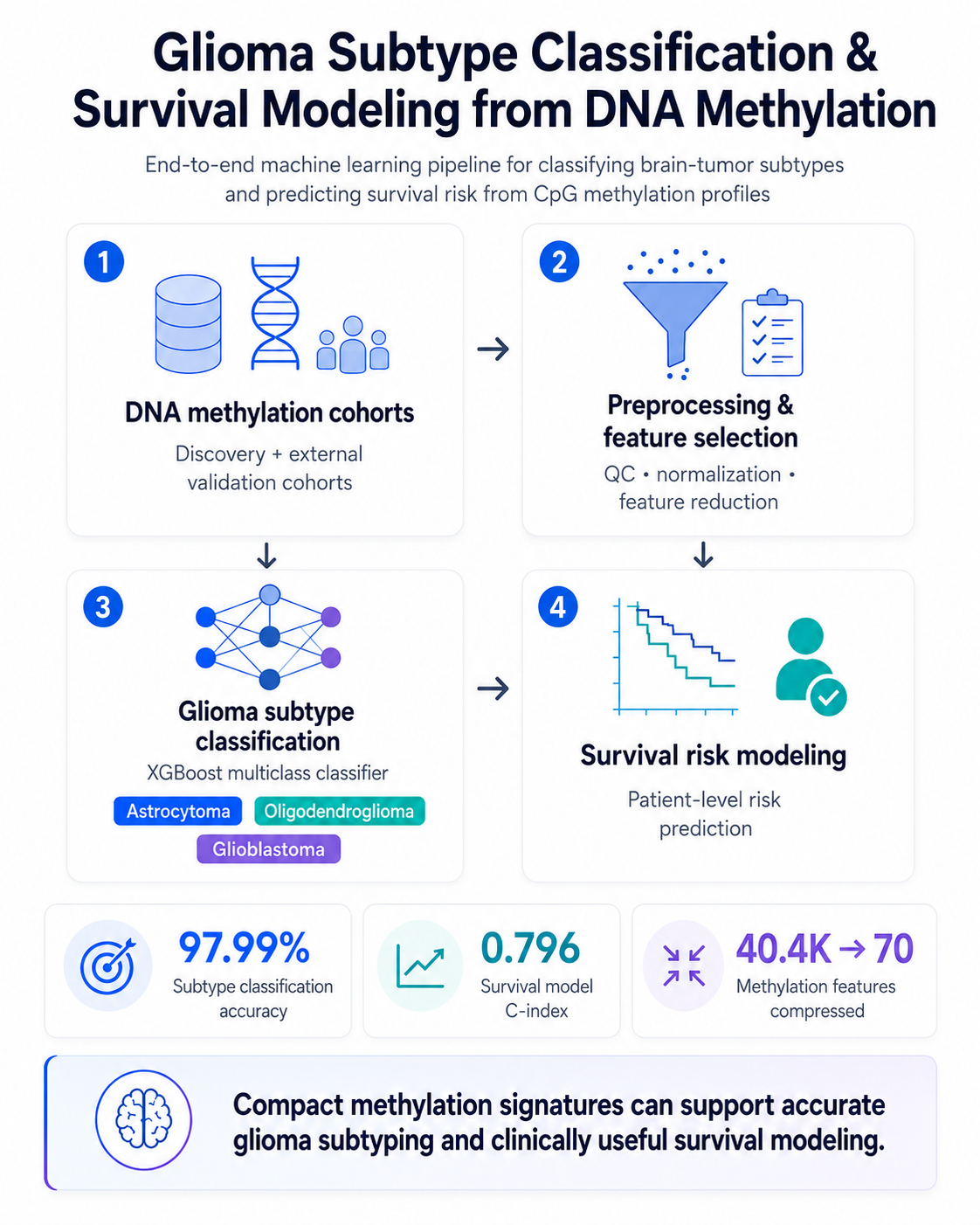
The problem
Glioma subtypes have very different survival outcomes, but standard methylation testing is slow and costly — out of reach for many clinics.
My contribution
Built an XGBoost classifier that compresses 404K methylation features to a 70-feature signature (97–99% accuracy), plus a Cox survival model (C-index 0.796) that surfaced novel prognostic markers.
Outcome
A compact 70-feature panel that could make subtyping faster and cheaper. Reproducible end-to-end pipeline; manuscript in progress with the Mamatjan Lab.
What I learned
Strict leakage control is the difference between optimistic-looking results and honest ones. SIS feature screening, scaling, and hyperparameter tuning must all be re-fit inside every training fold — otherwise the held-out folds aren’t really held out. The largest practical win came from gene-region aggregation: collapsing hundreds of thousands of CpGs to per-gene promoter and body summaries improved interpretability without sacrificing performance, and produced feature lists that biologists could actually engage with. Pooled-stratified modelling captured a single dominant cross-cancer risk axis with very few features, while per-cancer models surfaced richer histology-specific biology — different questions, different right tools.
- Type
- Model
- Role
- First author · ML lead
- Timeframe
- Feb — Jul 2025
- Stack
-
PythonRXGBoostglmnet (Elastic-Net Cox)CoxBoostscikit-learnpandasNumPyminfiBayesian optimization
- Tags
-
HealthMLBioinformaticsSurvival analysisGenomicsMethylation